

O título deste publish vem de um artigo publicado em uma edição digital especial sobre o tema “Química orgânica física: nunca fora de moda“(Cite) 10.1021/acs.joc.5c00426 (/cite) lá, Paul Rablen apresenta o caso de que a quantidade de o (Ortho) Produto em substituição eletrofílica de um anel fenil com um EWG (grupo de retirada de elétrons) é frequentemente grande o suficiente para merecer a mudança da regra de tempo há muito Esses substituintes são melhor compreendidos como orto, meta-diretores, com uma preferência por meta. Não posso deixar de acrescentar aqui uma citação (citação) 10.1039/ct8875100258 (/cite) à publicação mais antiga que posso encontrar mostrando tabelas de ambos O, p e m-Direcionando grupos e datados de 1887, então essa regra tem 138 anos (pelo menos).
Aqui, eu pensei que poderia mostrar alguns modelos computacionais (ωb97xd/def2-qzvpp/scrf = diclorometano) (cite) 10.14469/hpc/15341 (/cite) derivados da estabilidade relativa do possível ou σ-cúpula produzida pela protonação do ph-swg molecule para a tabela para explorar até que ponto esse efeito pode ser empurrado.
Começo analisando os resultados relatados para Benzonitrile (EWG = CN), para distribuições típicas de produtos:
- o– (~ 16%), m– (~ 82%) e p– (~ 2%) são citados para o íon nitronium como eletrofilo
- o– (23%), m– (74%) e p– (3%) para cloração
- o– (34%), m– (55%) e p– (1%) para brominação não catalisada (ver (cite) 10.1002/jcc.23985 (/cite) para um mecanismo inesperadamente complexo e análise cinética dessa reação em explicit)
- cálculos do complexo σ (citação) 10.1002/poc.4457 (/cite) que resultam em valores de o– (43%), m– (55%) e p– (2%) para benzonitrila.
- A observação foi feita (cite) 10.1002/poc.4457 (/cite) que A inclusão de uma correção de solvatação melhorou substancialmente o acordo com as informações experimentais limitadas disponíveis para nós sobre distribuições de produtos em EAS e os resultados abaixo certamente confirmam isso (especialmente para o benzonitrila). O solvente também tem um efeito significativo na geometria otimizada de cada sistema (consulte a tabela).
Os cálculos relatados aqui (cite) 10.14469/hpc/15341 (/cite) são semelhantes aos relatados usando um modelo ligeiramente diferente (cite) 10.1002/poc.4457 (/citação). Para o exemplo específico de benzonitrila, os autores do relatório authentic expressaram surpresa que seus cálculos mostraram que “Os complexos Ortho e Meta σ foram … sobre igualmente estáveis“Os resultados deste weblog mostram uma discriminação um pouco maior e talvez mais realista (?) Em favor de meta por 0,51 kcal/mol na energia livre.
Outras observações dignas de nota incluem isso
- Comparado com a CN, o grupo isonitrila iso-eletrônico NC é um forte e convencional o/p diretor, com uma preferência por p.
- O ewg r = bo (uma molécula conhecida, embora muito instável (cite) 10.1021/jo401942z (/cite)) é o próximo isômero isoeletrônico do CN e agora revela uma preferência muito forte pela meta-substituição, com apenas 3,5% de orto. Portanto, este grupo não segue a nova regra proposta de “orto, meta-diretores, com uma preferência por meta ” Embora seja improvável que isso seja capaz de ser testado experimentalmente devido à instabilidade dessa espécie (ela prontamente trimeriza).
- Finalmente, nessa progressão isoeletrônica para r = bef, os cálculos parecem agora mostrar que este é um forte o– Diretor (61%) e que m é apenas 29%, novamente não seguindo a regra recém -modificada, mas provavelmente não testável.
- R = no entanto, no entanto, parece ser um exemplo da nova regra modificada, pois a porcentagem de o– é tão alto quanto 23,8%. Aqui é significativo que tanto para o– e m– σ complexos, O grupo NO foi calculado como sendo co-planejado com o anel fenil, indicando uma conjugação significativa-mas o p-Imômero (2,3%) foi torcido e, portanto, não conjugado (valores diédricos mostrados abaixo).
- O mesmo resultado é obtido para r = não2com o p-Isômero com um ângulo de torção de 67 °.
| Intermediários catiônicos em substituição eletrofílica de ph-r | ||||||
|---|---|---|---|---|---|---|
| R | ΔΔG298Kcal/mol (pop, %) Ortho, |
rCr UM |
ΔΔG298Assim, (pop, %) Meta |
rCr | ΔΔG298Assim, (pop, %) par |
rCr |
| NC, gás | -4,72 (21.42) |
1.349 | 0,0 (0,01) |
1.369 | -5,51 (78,57) |
1.348 |
| NC, DCM | -2,50 (35.51) |
1.359 | 0,0 (0,56) |
1.377 | -2.86 (63,93) |
1.359 |
| CN, gás | -1.38 (60,56) |
1.423 | 0,0 (6.07) |
1.433 | +0,36 (33.37) |
1.425 |
| CN, DCM | +0,51 (27.68) |
1.428 | 0,0 (64,05) |
1.435 | +1.23 (8.27) |
1.433 |
| Bogás | +0,96 (16,76) |
1.541 | 0,0 (82,34) |
1.540 | +2.72 (0,09) |
1.549 |
| BoDCM | +1.99 (3.52) |
1.537 | 0,0 (96,34) |
1.532 | +3.93 (0,14) |
1.547 |
| Bef, gás | +0.23 (38,78) |
1.727 | 0,0 (56,73) |
1.714 | +1.53 (4.49) |
1.737 |
| Bef, dcm | -0,46 (61.21) |
1.748 | 0,0 (28,66) |
1.731 | +0,63 (10.13) |
1.762 |
|
|
||||||
| Cf.3gás | +0,25 (30,86) |
1.524 | 0,0 (46,87) |
1.521 | +0,45 (22.27) |
1.533 |
| Cf.3DCM | +1.45 (8.11) |
1.518 | 0,0 (89,66) |
1.513 | +2.22 (2.23) |
1.528 |
| Não, gás | +0.44 (25.07) |
1.460 | 0,0 (52,32) |
1.477 | +0,51 .22.61) |
1.395 |
| Não, DCM | +0,68 (23,84) |
1.458 | 0,0 (73,87) |
1.456 | +2.09 (2.29) |
1.429 |
| NÃO2gás | +1.08 (13.38) |
1.487 | 0,0 (79,88) |
1.487 | +1.49 (6.73) |
1.476 |
| NÃO2DCM | +1.80 (4.73) |
1.480 | 0,0 (94,25) |
1.478 | +2.73 (1,01) |
1.481 |
Para a explicação sugerida, (cite) 10.1021/acs.joc.5c00426 (/cite) onde a interação dos elétrons π do complexo σ com o π* orbital do ewg foi sugerido para ser mais forte não apenas para o M-Ersomer, mas também o O-isomer como PJ-IM para que, em comparação. Isso agora pode ser quantificado usando Análise NBO7que indica a energia de interação entre pares de doadores preenchidos e orbitais de aceitadores vazios.
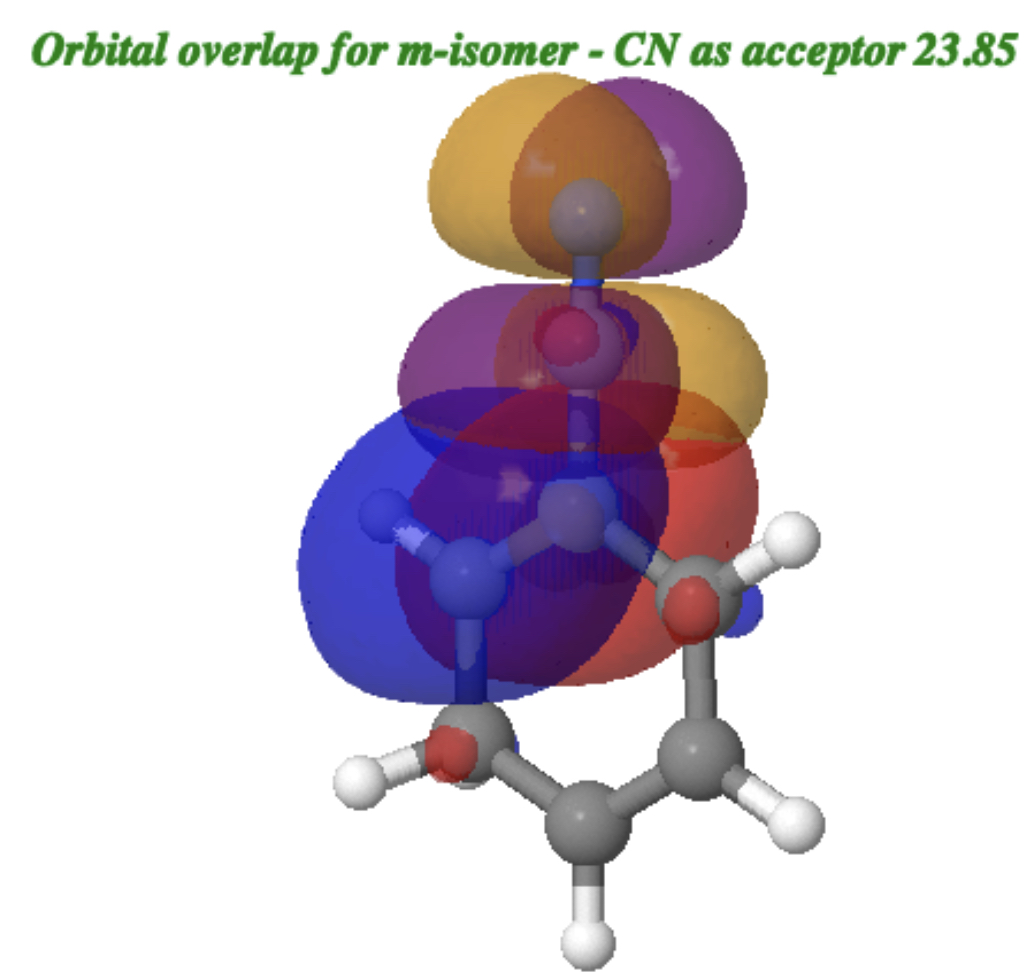
Para o isômero m (citação) 10.14469/hpc/15354 (/cite) de benzonitrila protonada, a sobreposição dos dois orbitais é mostrada abaixo (clique na imagem para obter um modelo 3D rotativo) com o azul positivo positivamente com roxo e vermelho com laranja. A energia de interação NBO E (2) é de 23,85 kcal/mol.
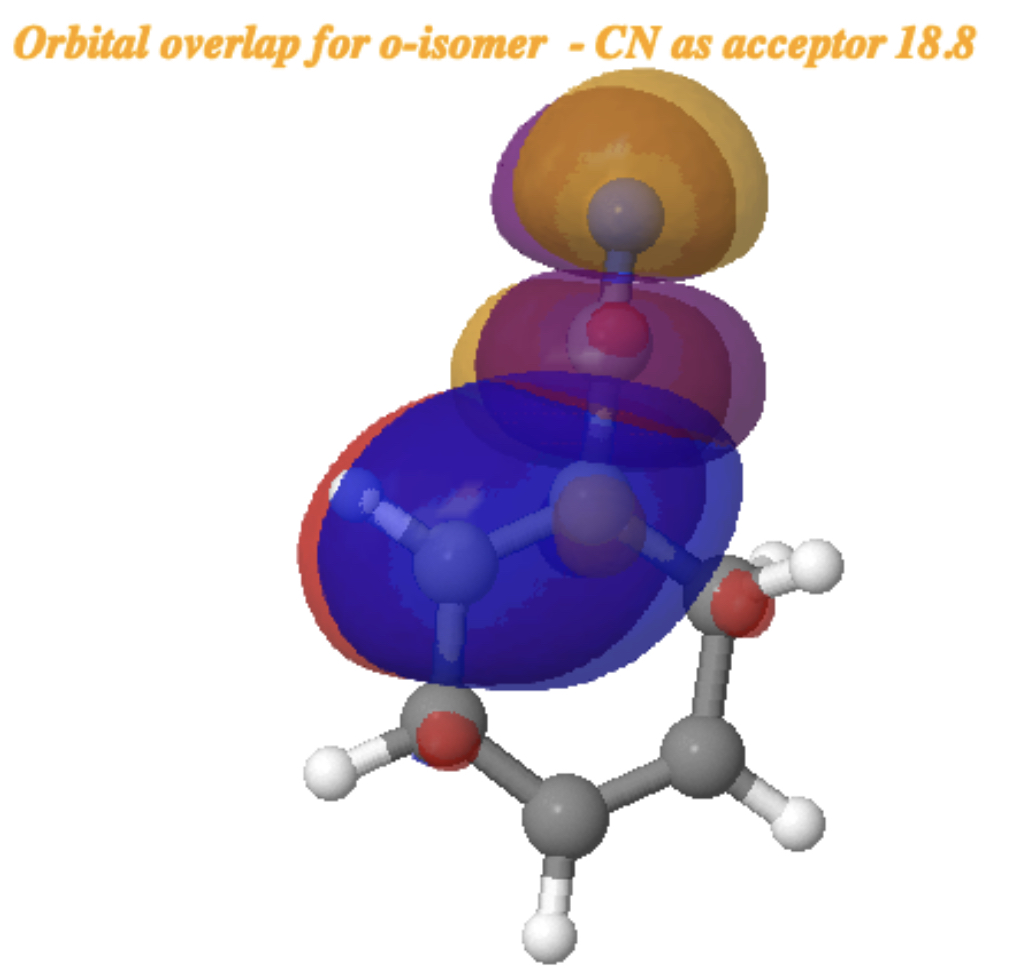
Para o o-Isômeros (citação) 10.14469/hpc/15355 (/cite) (abaixo), a energia de interação NBO E (2) é um pouco reduzida para 18,8 kcal/mol, mas ainda é considerável e mais ou menos proporcional às energias livres relativas da o– e m-Isômeros.
No entanto, para o p-Isômero (citação) 10.14469/hpc/15353 (/cite), a sobreposição orbital equivalente é mostrada abaixo; Não possui interação actual e, portanto, o E (2) é efetivamente muito pequeno.
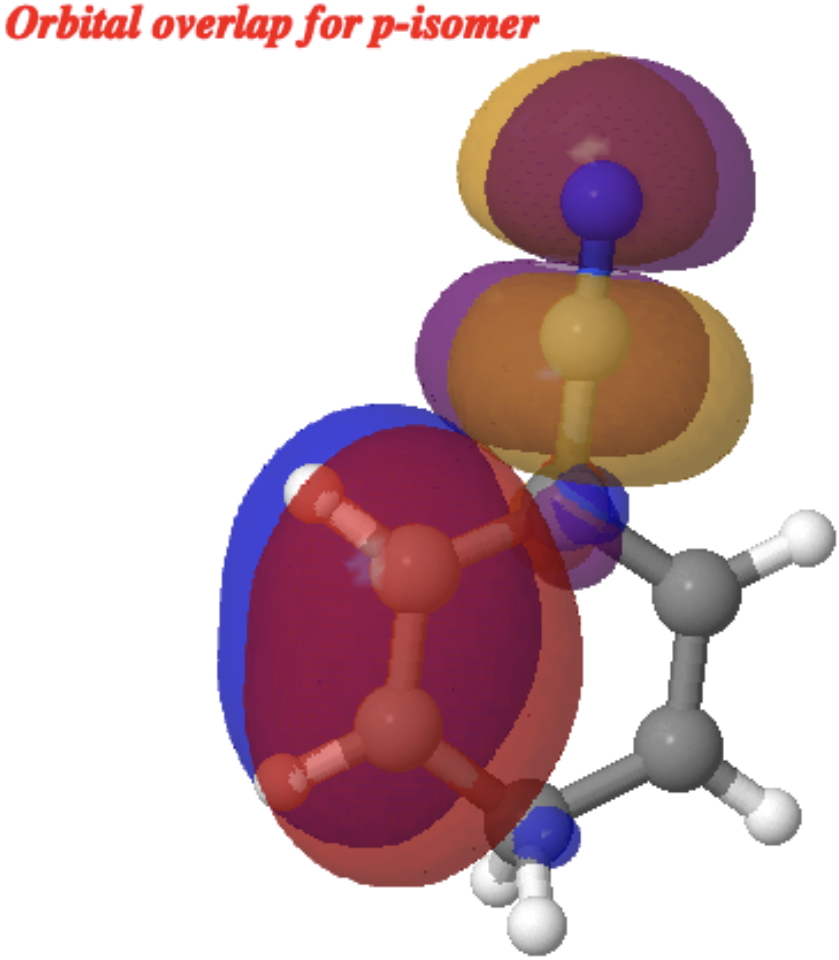
Além disso, a energia de interação NBO E (2) para os mesmos átomos que com o– e m– mostra duas instâncias de 3,0 kcal/mol (por causa do C2V simetria), muito reduzida dos outros dois isômeros.
Embora muitas outras interações possam ser encontradas na análise da NBO, isso explica de longe a maior diferença entre o oAssim, me p isômeros. Esses resultados também correspondem à observação feita acima daquele para r = não, o o– e m-Imômeros são totalmente coplanares, mas para o p-Imômeros O grupo NO é torcido em cerca de 90 ° em relação ao anel fenil. Isso também se reflete nas vibrações de torção ou torção calculadas do grupo R, sendo 89 cm-1 para m-Nitroso vs. 23 cm-1 para o-nitroso e novamente 55 cm-1 para m-nitro vs. 38 cm-1 para o-nitro.
Portanto, esta nova análise de sobreposição orbital NBO7 ajuda a quantificar esses efeitos (a análise qualitativa relatada (citação) 10.1021/acs.joc.5c00426 (/cite) foi baseada em orbitais moleculares em vez de orbitais da NBO localizados) e confirma que, pelo menos, para alguns grupos EWG, o que o-Imômeros é quase tão favorecido quanto o m-forma. Bem, uma observação que tem 138 anos recebe uma nova luz sopra!
Este publish tem doi: https://doi.org/10.59350/rzepa.28993
Relacionado
Esta entrada foi publicada na quinta -feira, 17 de julho de 2025 às 12:44 e é arquivada em Química interessante. Você pode seguir qualquer resistência a esta entrada através do RSS 2.0 alimentar. Você pode Deixe uma respostaou trackback do seu próprio web site.




")

{kind=link}