

A cristalografia de raios-X é a técnica de usar a difração de raios X pelos elétrons em uma molécula para determinar as posições de todos os átomos nessa molécula. A teoria quântica nos ensina que os elétrons podem ser encontrados em conchas ao redor dos núcleos atômicos. Existem dois tipos amplos, a concha mais externa (também chamada de concha de valência) e todas as conchas internas ou principais. A densidade dos elétrons do núcleo é muito maior (mais compacta) do que a concha de valência mais difusa para todos, exceto o átomo de hidrogênio, que possui apenas elétrons de valência. Como isso se relaciona com a difração de raios-X por elétrons? Bem, os principais elétrons, devido à sua relativa compactação, difratar raios X mais fortemente do que os elétrons de valência. Essa compactação do núcleo também significa que sua distribuição de densidade de elétrons pode ser bem (mas não exatamente) aproximada por uma esfera, com o núcleo no centro dessa esfera. E a partir disso segue que a densidade para cada átomo pode ser tratada de forma independente, o chamado modelo IAM ou átomo independente. Por exemplo, todos os átomos de carbono em uma molécula são aproximados como tendo o mesmo valor para a densidade de elétrons da concha principal. Mas a aproximação do IAM é muito menos boa para os átomos de hidrogênio, especialmente quando são ligados a átomos muito polares (Li, O, F, and many others) e até átomos como carbono ou oxigênio têm desvios perceptíveis, conforme ilustrado na Figura 1 abaixo. (cite) 10.1039/d0sc05526c (/cite)
Figura 1 De (cite) 10.1039/d0sc05526c (/cite) com legenda: Densidades de deformação hirshfeld para os átomos de carbono (esquerda) e oxigênio (direita) no grupo carboxilato de Gly-L-Ala, ou seja, diferença entre a densidade de elétrons atômicos esféricos utilizada no refinamento do IAM e do não-esférico hirshfeld usado no refinamento de hirshfeld = har (ioteca (izeado (ioteca). Vermelho = negativo, azul = positivo. Isovalue = 0,17 eå-3.‡
A cristalografia de raios-X tem tudo a ver com corresponder o mapa de densidade de elétrons de uma estrutura de modelo com o mapa de densidade de elétrons derivado dos dados de difração. Na cristalografia de raios X “convencional”-ou seja, usada pela maioria dos cristalógrafos-o mapa de densidade de elétrons do modelo é calculado usando a abordagem IAM, onde nenhuma consideração é dada a qualquer distorção da distribuição de densidade de elétrons causada por coisas como ligações-cada átomo é tratado independentemente (portanto, o nome). Esse método luta especialmente com hidrogênios e, portanto, a posição inferida do núcleo de hidrogênio no centro de uma distribuição esférica assumida é frequentemente difícil de obter com precisão. Digite a cristalografia quântica, pela qual um modelo de distribuição de densidade de elétrons em uma molécula pode ser calculado resolvendo a equação de Schrodinger, hoje em dia com uma aproximação muito razoável em um tempo razoável (minutos) usando a chamada teoria funcional da densidade, ou DFT. Pode -se esperar que o mapa de densidade de elétrons resultante para a estrutura do modelo corresponda a realidade mais de perto do que a abordagem do IAM. O mais obviamente afetado por essa mudança é o manuseio de átomos de hidrogênio. Se alguém considera uma ligação C -H de um SP3 O átomo de carbono, usando uma aproximação do IAM, o átomo de hidrogênio (ou seja, seu núcleo ou próton) seria colocado no centro de densidade máxima de elétrons, no pleno conhecimento de que não é aqui que está o próprio núcleo do átomo de hidrogênio. A direção do vetor C -H estaria correta, mas a distância seria muito curta. Na abordagem de cristalografia quântica, as posições dos núcleos de átomos de hidrogênio, por exemplo, não são exatamente coincidentes com os máximos de densidade de elétrons, totalizando os átomos não esféricos, evitando assim os erros sistemáticos vistos na abordagem do IAM. Menores, mas possivelmente ainda significativos, esses erros podem ser esperados para os elementos da segunda linha e além.
A obtenção de posições confiáveis de átomos de hidrogênio exigia anteriormente um estudo de difração de nêutrons, que é difícil, caro e demorado. Portanto, a idéia de usar as densidades DFT não esféricas, em vez da abordagem esférica do IAM para construir um modelo usando dados de difração de raios-X, é muito atraente. Mas funciona? Para testar isso, decidimos voltar a algumas estruturas publicadas anteriormente que foram tratadas usando a abordagem IAM e refinando-as usando cristalografia quântica. Não temos os correspondentes estudos de nêutrons para verificar as respostas, mas ainda podemos ver o quão bem as próprias estruturas refinam e que novos problemas essa abordagem pode surgir.
Método
As estruturas publicadas originais (cite) 10.14469/hpc/2297 (/cite) foram refinadas com Shelx-2014 (cite) 10.1107/s2053229614024218 (/cite) que utiliza um modelo de átomo independente (IAM). Os resultados relatados aqui empregados Noshera2(cite) 10.1039/d0sc05526c (/cite), (cite) 10.1107/s0021889808042726 (/cite) usando Refinamento de átomos de Hirshfeld(cite) 10.1107/s2052252514014845 (/citar) e selecionar DEF2-SVP como o conjunto de bases (todos os átomos)† e ωB97X-V como o método DFT (os resultados parecem relativamente insensíveis a qualquer um), implementados no programa ORCA. (CITE) 10.1002/WCMs.81 (/citação) para as primeiras tentativas não foram feitas alterações nas estruturas além do refinamento anisotrópico dos agora átomos de hidrogênio sem restrições. Para quatro das estruturas, vários átomos de hidrogênio foram definidos não positivos (ou seja, um dos raios de um elipsóide térmico refinado a um comprimento negativo), que é fisicamente sem sentido e seria uma barreira significativa à publicação. (Não queremos dizer “inadequados”, pois quase sempre há exceções, mas um parâmetro térmico definido não positivo é bem próximo de ser inaceitável.) Para esses casos, foi criada uma segunda versão (V2), onde todos os átomos de hidrogênio foram relevados isotropicamente, mas com as distâncias e parâmetros térmicos ainda permitidos para refinar. Para AB1709 (18b), isso ainda possuía o parâmetro térmico isotrópico de um dos átomos de hidrogênio (H11) GO não positivo definido; portanto, para aquele átomo de hidrogênio, o parâmetro térmico isotrópico livre foi substituído por uma pilotagem.
Os resultados
Escolhemos um conjunto de sete estruturas publicadas em 2017 (cite) 10.1021/acomega.7b00482 (/cit) e refinadas conforme observado acima usando métodos convencionais. Esses sete também compreendem um dos primeiros conjuntos de estruturas cristalinas para as quais os dados completos de difração foram disponibilizados (citação) 10.14469/hpc/2297 (/cite) em vez de apenas a estrutura refinada na forma de um arquivo CIF. Os novos resultados também foram depositados (cite) 10.14469/hpc/15030 (/cite) para aumentar o registro desses compostos. As planilhas correspondentes às imagens abaixo podem ser obtidas clicando na imagem.
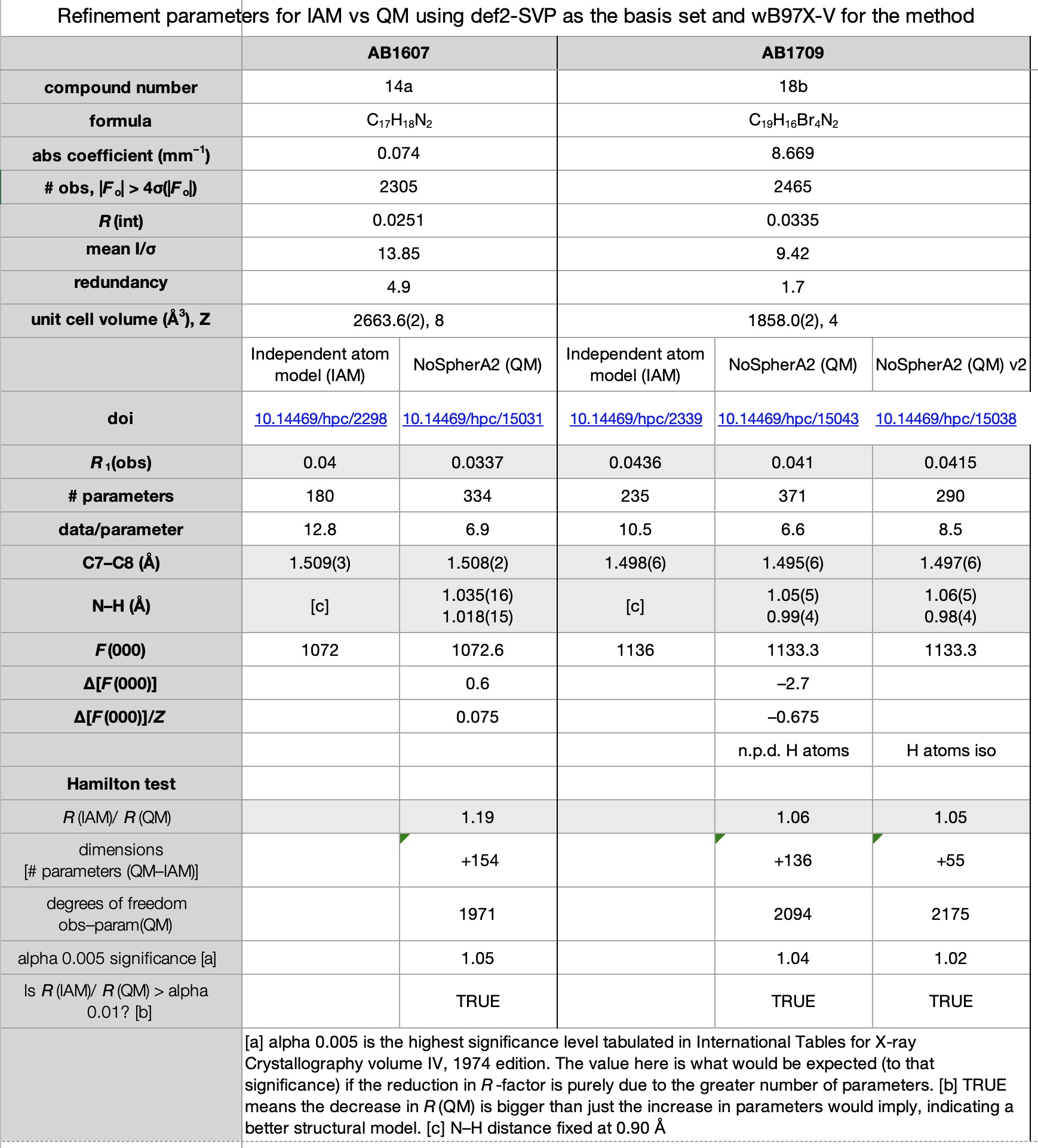
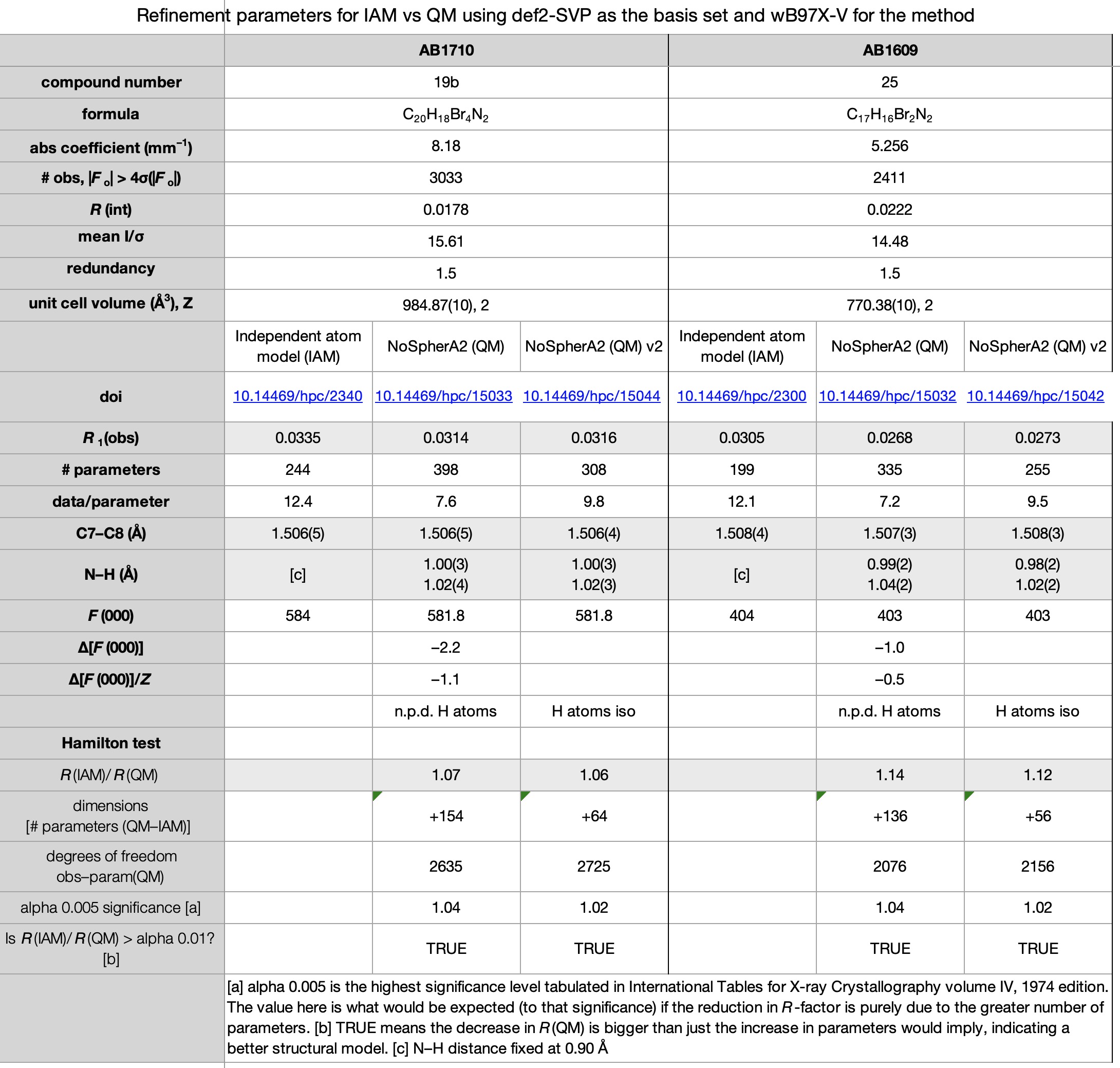
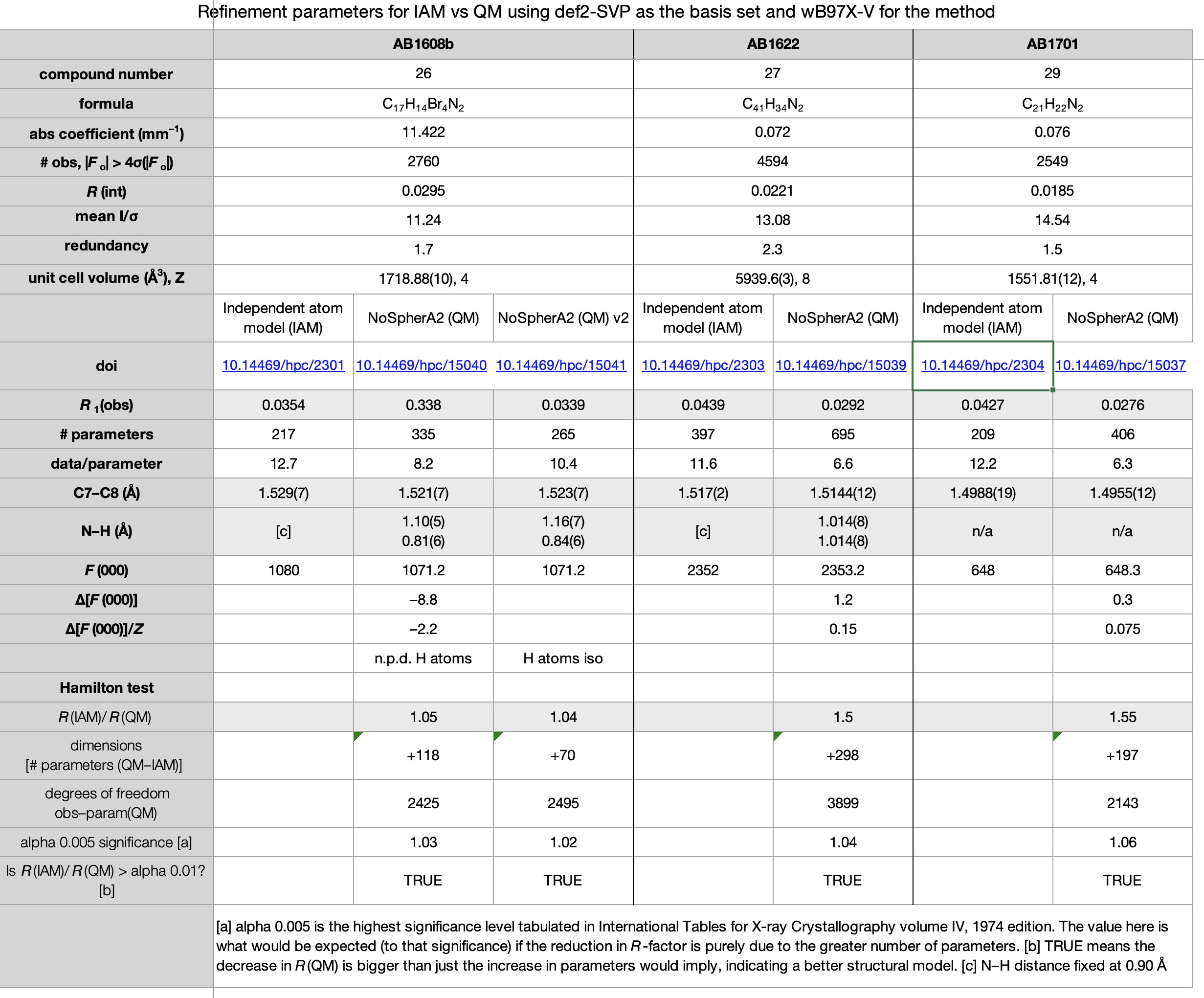
- Todas as sete estruturas viram uma redução no fator R last. (CITE) 10.14469/hpc/15030 (/cite) No entanto, todas as estruturas também viram um aumento significativo no número de parâmetros (como os átomos de hidrogênio passaram do uso de zero parâmetros de cada modelo de reinício para nove parâmetros de um pouco livre de um livre e totalmente livre. No entanto, todos os refinamentos de QM passaram o Teste de Hamiltonsugerindo que os fatores R reduzidos de fato refletir um modelo melhorem vez de apenas ser uma conseqüência do número significativamente aumentado de parâmetros.
- Todas as quatro estruturas contendo átomos de bromo tiveram vários átomos de hidrogênio não positivos quando refinados anisotropicamente. Não está claro exatamente por que isso aconteceu-não parece haver nenhuma correlação com a qualidade ou intensidade dos dados (conforme medido de maneira grosseira por r (int) e média I/σ, respectivamente) e, embora a redundância dessas estruturas seja bastante baixa (entre 1,5 e 1,7), aqueles para as estruturas não-brominos não são muito melhores (1.5, 2.3 e 4.9). Esses conjuntos de dados foram o resultado de experimentos projetados para coletar 98,5% dos dados exclusivos da simetria, sem considerar a redundância. No entanto, a comparação das versões iniciais e secundárias dos refinamentos dessas quatro estrutura mostra que a maioria substancial da diminuição do fator R observada pode ser alcançada sem o uso de átomos de hidrogênio anisotrópico.
- No que diz respeito à precisão das estruturas, usando uma ligação C (Sp2) –C (SP3) como proxy (a ligação C7 -C8), podemos ver que o desvio padrão estimado é o mesmo ou apenas ligeiramente menor nas sete estruturas, sugerindo que a obtenção de ESDs mais baixos não seria um fator motivador para o uso de cristalografia quântica.
- Um dos resultados mais inesperados foi a variação em F (000). Na cristalografia de raios X (ênfase deliberada no raio-x, pois a difração de nêutrons é diferente) F (000) deve ser o número complete de elétrons presentes na célula unitária e é usado como um fator de escala geral para o mapa de densidade de elétrons. É muito não supostamente variável, e qualquer discrepância indicaria um erro na fórmula calculada ou relatada e deve ser corrigida. Não entendemos por que o Os refinamentos de QM dão uma resposta diferente dos do IAM (Alguns e outros para baixo-normalizados para uma base por molécula, o intervalo é –1,1 a +2,2), embora pareça provável que esteja associado a pontos de corte (limites) na medição dos valores de elétrons “manchados” nos modelos QM, todos os modelos de IAM fornecem os valores “corretos”.
- Com base nos relatórios do CheckCIF para as estruturas QM, se a cristalografia quântica capturar em grande parte, o checkcif provavelmente precisará ser atualizado, agora haverá vários alertas de alto nível para ligações X -H longas.
- Uma das principais áreas de incerteza com cristalografia quântica é o que/quanto os dados precisam ser coletados. Dados exclusivos de simetria para 0,84 Å parecem insuficientes, mas o que seria suficiente – esfera completa, redundância, maior resolução? Os resultados finais valeriam o investimento de tempo additional? Nenhum dos aspectos acima é claro nesta fase, mas será interessante ver como a técnica se desenvolve.
Essas sete estruturas cristalinas também ocupam uma posição interessante para a posteridade. Os dados para eles foram disponibilizados em oito anos, o que ilustram dois métodos de refinamento significativamente diferentes usados durante esse período, além de ter acesso aos dados de imagem de difração completos originais para permitir que qualquer análise completamente nova seja feita no futuro. Quem sabe, talvez daqui a oito anos um método ainda melhor possa estar disponível para comparação com os resultados relatados aqui.
‡Para colocar isso em contexto, 0,17 ea-3 Geralmente seria considerado um ruído de fundo bastante baixo, semelhante ao valor da densidade de elétrons residuais máximos com a qual um cristalógrafo poderia estar feliz. †A estrutura que mostrou a menor mudança no fator R no uso de cristalografia quântica, ou seja O AB1608B, foi re-executado com o conjunto de bases triplo-ζ def2-tzvpp. Isso deu fatores R mais baixos, mas muito pouco (3,38% a 3,36% ANISO com NPD; 3,39 a 3,38 ISO).
Relacionado
Esta entrada foi publicada na segunda -feira, 17 de março de 2025 às 19:57 e é arquivada em Química interessante. Você pode seguir qualquer resistência a esta entrada através do RSS 2.0 alimentar. Você pode Deixe uma respostaou trackback do seu próprio web site.




")

{kind=link}