

Geometrias de transferências de prótons: modeladas usando energia whole ou energia livre?
As transferências de prótons estão entre as mais comuns de todas as reações químicas. Eles são frequentemente considerados como “triviais” e podem não aparecer em muitos esquemas mecanicistas, exceto talvez a notação “pt”. Os tipos com as mais baixas barreiras de energia para transferência geralmente envolvem heteroátomos como N e O, e o estado de transição convencional pode ser quando o próton estiver localizado na distância de meio caminho entre os dois heteroátomos. Este deve ser o ponto alto da energia entre as duas posições para o próton. Mas e se uma estrutura cristalina for determinada com o próton exatamente nessa posição? Bem, a primeira hipótese é que o uso de raios-X como radiação difradora não é confiável, porque os prótons dispersam os raios-X muito mal. Então, às vezes, é realizado um estudo de difração de nêutrons mais árduos, o que geralmente se supõe ser mais confiável na determinação da posição do próton. Exas assim esse estudo foi realizado para a estrutura mostrada abaixo (Rakqoj) (cite) 10/c3zxh2 (/cite), Datadoi: 10.5517/cc57db3 Para a determinação de 80k. Os substituintes foram selecionados para tentar maximizar a simetria do O… H… n Motif by way of Ajuste da PKA (para outra tentativa de ajuste, Veja este weblog). A paisagem mais geral que essa molécula se encaixa (cita) 10.1039/c1ra00219h (/cite) é mostrada abaixo:
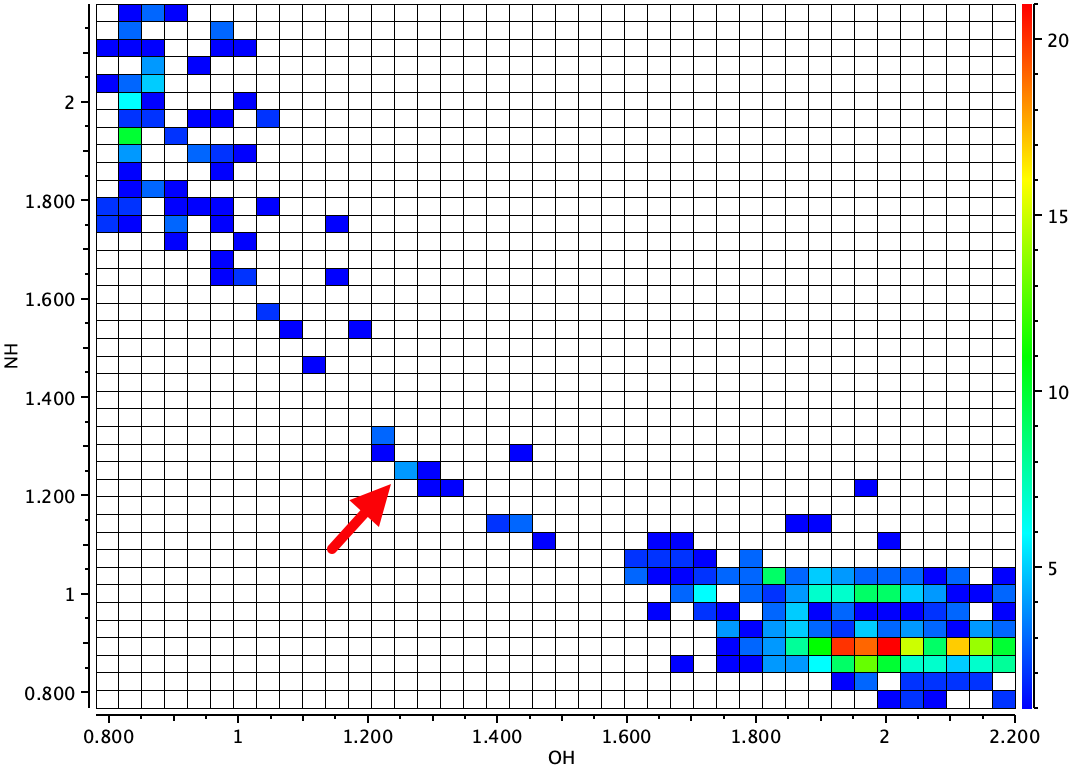
Os resultados obtidos para a posição do próton para Rakqoj foram fascinantes. Eles eram muito dependentes da temperatura do cristal! Em temperaturas da sala (usando raios-X), o próton foi medido como 1,09å do oxigênio e 1,47å da nitrogênio (forma neutra acima). A 20K, a distância OH foi de 1,309å e o HN 1.206å (~ forma iônica acima). De fato, o próprio título deste artigo é Primeira ligação OHN de hidrogênio com um próton centrado obtido por migração de prótons induzida termicamente. Os autores dão várias razões para esse comportamento (seu ref 17 (cite) 10/c3zxh2 (/cite) e também (cite) 10.1039/c1Ra00219h (/cite)), mas um que eles não mencionam são alterações térmicas induzidas no The the Constante dielétrica do cristal com temperatura, dado que em uma posição para o próton, a molécula é iônica e no outro neutro. Então eu decidi modelar o sistema em função do solvente. Neste modelo, o dielétrico do solvente é usado para aproximar o dielétrico cristalino. Minha primeira escolha de função de energia é calcular geometrias usando o b3lyp+gd3bj/def2 = tzvpp/scrf = método solvente para ver o que pode emergir e como um possível prelúdio para experimentar outros funcionais. Dados justos para esses cálculos são coletados no DOI: 10.14469/hpc/10368.
| Solvente | ε | ΔG298 para o… hn | rOH | rHn | ΔG298 para oh… n | rOH | rH… n | ΔG298 TS (PT) |
rOH | rHn |
|---|---|---|---|---|---|---|---|---|---|---|
| Água | 78.4 | -2893.387188 -2893.334325♠ |
1.4913 | 1.0827 | -2893.386705 -2893.334333♠ |
1.0364 | 1.5696 | -2893.387668 -2893.336183♠ |
1.1852 | 1.2899 |
| Dicloro metano |
8.9 | -2893.385173 | 1.4566 | 1.0945 | -2893.385662 | 1.0309 | 1.5878 | -2893.386022 | 1.2072 | 1.2642 |
| Clorofórmio | 4.7 | -2893.382254 | 1.4227 | 1.1082 | -2893.384514 | 1.0261 | 1.6049 | -2893.384773 | 1.2321 | 1.2388 |
| Éter dibutil | 3.1 | -2893.380813 | 1.3778 | 1.1302 | -2893.383511 | 1.0213 | 1.6235 | -2893.382918 | 1.2667 | 1.2078 |
| Tolueno | 2.4 | -2893.379752 | 1.3248 | 1.1635 | -2893.382915 | 1.0178 | 1.6385 | -2893.379773 | 1.2851 | 1.1934 |
| Fase gasosa | 0 | n / D | -2893.377949 | 1.0009 | 1.7387 | n / D | ||||
| Expt (RT) (cite) 10/c3zxh2 (/cite) |
? | n / D | 1.09 | 1.47 | n / D | |||||
| Expt (20k) (cite) 10/c3zxh2 (/cite) |
? | n / D | 1.309 | 1.206 | n / D | |||||
♠ Em 20k
Resultados:
- As geometrias para cada modelo são obtidas minimizando o energia whole do sistema em função das variáveis geométricas 3N-6 (coordenadas).
- As geometrias mostram que, para todos os solventes, dois mínimos na energia whole são obtidos, um para o iônico e outro para a forma neutra. Isso é chamado de potencial de energia de poço duplo. Mesmo um solvente não polar, como o tolueno, produz uma energia de solvatação de ~ 3,1 kcal/mol em comparação com a fase gasosa, o que é suficiente para induzir um potencial de poço duplo.
- Sem solvente (fase gasosa), apenas a geometria neutra é obtida.
- Na água de solvente mais polar, o potencial duplo de poço se parece com o seguinte:

O poço iônico é de cerca de 0,4 kcal/mol menor em energia whole (e ~ 0,3 kcal/mol em energia livre, consulte a tabela acima) do que a forma neutra, com uma barreira conectando neutro a iônico apenas 1,0 kcal/mol. Um estado de transição + coordenada de reação intrínseca (IRC) pode ser facilmente localizada nesse potencial whole de energia, confirmando a forma de poço duplo. - Quando as energias livres ΔG são calculadas, que incluem efeitos térmicos, como entropia e energia de ponto zero, o estado de transição surge como 0,3 kcal/mol Menos que a energia whole da forma iônica (entradas vermelhas, tabela). Com efeito, a superfície do potencial de energia livre é invertida em comparação com a superfície whole de energia e o “estado de transição” se torna o ponto mais baixo na superfície de energia. Portanto, esse ponto é um mínimo na energia livre, mas um máximo na energia whole, o resultado da adição de efeitos térmicos à energia whole.
- No diclorometano, a energia livre da forma neutra agora é menor em 0,3 kcal/mol que a forma iônica. O OH Bond está começando a ficar mais curto e o NH um mais longo. O estado de transição agora é 0,22 kcal/mol menor que a forma neutra. Com o clorofórmio, as ligações OH e HN se tornaram ~ igual em comprimento, o próton é descartado simetricamente.
- Quando o éter dibutil como solvente é atingido, o estado de transição não é mais menor em ΔG do que a forma neutra, passando a ser 2,0 kcal/mol mais alto para tolueno. Assim, à medida que a polaridade do solvente diminui, vemos uma alteração no potencial de um único poço em ΔG, no qual o próton é centrado, para um poço muito assimétrico no qual o próton está ligado ao oxigênio.

- Podemos corresponder aos resultados de difração de nêutrons observados com os cálculos? À medida que a temperatura diminui, a difração de nêutrons mostra o início da transferência de prótons do oxigênio para o nitrogênio para formar uma espécie iônica. Os cálculos mostram que isso pode ser modelado por um aumento na constante dielétrica efetiva do meio. O “estado de transição” computado para a transferência de prótons em algum lugar entre éter dibutil e tolueno (como um meio dielétrico) surge como aproximadamente o melhor modelo para a estrutura dessa espécie. Nesse dielétrico, o ΔG calculado não é mais o menor ponto de energia livre no potencial. Isso pode ser devido às muitas aproximações usadas neste modelo, como a minimização da energia whole, o método da função de partição usado para calcular a entropia, a natureza da DFT funcional, o modelo de solvatação contínua, o conjunto de bases, and so on.
Conclusões:
Esses resultados foram obtidos com a aproximação de que minimizando a energia molecular whole produz uma geometria computada que pode ser comparada às estruturas experimentais de difração de nêutrons. Mas pode -se fazer melhor? A obtenção de geometrias moleculares, minimizando as energias livres calculadas, seria não trivial. Em primeiro lugar, a minimização dependeria da disponibilidade de primeiros derivados da função energética em relação às coordenadas, neste caso ΔG. Estes não estão disponíveis para nenhum código DFT. O resultado seria dependente da temperatura (como de fato são os resultados experimentais mostrados acima). Além disso, ΔG é calculado a partir de modos vibracionais normais e estes são apropriados apenas quando os primeiros derivados da função são zero; nesse momento, as chamadas seis rotações e traduções da molécula no espaço livre também têm energia zero. Portanto, precisamos de vibrações para calcular derivados, mas precisamos de derivados para calcular vibrações nessa abordagem clássica.
Seria ótimo, por exemplo, se o modelo aproximado do potencial para uma transferência de hidrogênio usado acima, com base na minimização do whole de energias para derivados, pudesse ser verificado em um modelo baseado em geometrias otimizadas usando energias livres. Tais procedimentos existem, (cite) 10.1063/1.2715941 (/cite) usando métodos de trajetória de dinâmica molecular.
Este put up tem doi: 10.14469/hpc/10382 (cite) 10/hqsm (/cite)
Relacionado
Esta entrada foi publicada na segunda -feira, 18 de abril de 2022 às 19:09 e é arquivada em cristal_structure_miningAssim, mecanismo de reação. Você pode seguir qualquer resistência a esta entrada através do RSS 2.0 alimentar. Você pode Deixe uma respostaou trackback do seu próprio website.




