

Conceitos Básicos
Este artigo discute o sequenciamento de proteínas e suas técnicas e aplicações na ciência moderna.
Tópicos abordados em outros artigos
Introdução ao sequenciamento de proteínas
O sequenciamento de proteínas é o processo de determinação da ordem precisa de aminoácidos em um proteína. As proteínas, compostas por longas cadeias de aminoácidos, são fundamentais para quase todos os processos biológicos. As proteínas derivam grande parte de sua função de seu estrutura tridimensionalque é determinado pela sequência de aminoácidos e como a proteína dobras. Portanto, compreender a sua sequência é essencial, pois mesmo pequenas mudanças na sequência podem alterar drasticamente a sua função. As sequências de proteínas oferecem informações vitais nas áreas de desenvolvimento de medicamentos, diagnóstico de doenças e biologia evolutiva.
Métodos Tradicionais de Sequenciamento de Proteínas
As técnicas tradicionais de sequenciamento de proteínas lançaram as bases para a proteômica moderna e forneceram insights críticos sobre a estrutura primária das proteínas. Dois métodos tradicionais de sequenciamento de proteínas incluem degradação de Edman e sequenciamento baseado em espectrometria de massa.
1. Degradação de Edman
Uma das primeiras e mais amplamente utilizadas técnicas é Edman degradaçãodesenvolvido por Pehr Edman na década de 1950. A degradação de Edman take away sequencialmente um aminoácido de cada vez do terminal N de uma proteína. Isto permite que a sequência de aminoácidos seja determinada passo a passo. O aminoácido clivado é então rotulado e identificado, enquanto a cadeia peptídica restante sofre novos ciclos de degradação até que toda a cadeia seja identificada.
Este método baseia-se na utilização de fenilisotiocianato (PITC) para reagir com o grupo amino livre do aminoácido N-terminal sob condições levemente alcalinas. A reação forma um derivado de feniltiocarbamida (PTC). Este é então clivado da cadeia peptídica como um derivado da tiazolinona, deixando o peptídeo restante intacto. A tiazolinona é convertida em um derivado de feniltiohidantoína (PTH) mais estável, que pode ser identificado usando técnicas cromatográficas, como cromatografia líquida de alta eficiência (HPLC). Embora altamente eficaz para peptídeos mais curtos (até cerca de 50 aminoácidos), a degradação de Edman tem limitações com sequências mais longas, proteínas com terminais N bloqueados ou resíduos modificados.
2. Sequenciamento baseado em espectrometria de massa
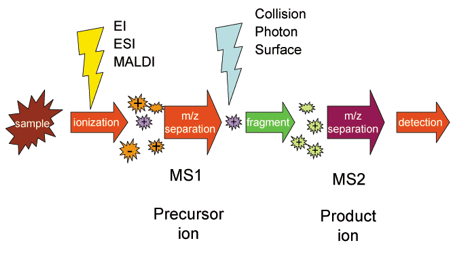
Espectrometria de massa (MS)O sequenciamento de proteínas baseado em proteínas revolucionou o campo da proteômica devido à sua velocidade, sensibilidade e capacidade de analisar misturas complexas de proteínas. Neste método, as proteínas são primeiro digeridas em peptídeos menores, normalmente usando enzimas como a tripsina. A tripsina cliva a proteína em resíduos de aminoácidos específicos para produzir fragmentos peptídicos previsíveis. Os fragmentos peptídicos resultantes são então ionizados, usando técnicas como ionização por eletrospray (ESI) ou dessorção/ionização por laser assistida por matriz (MALDI). Uma vez ionizados, esses peptídeos são introduzidos no espectrômetro de massa, onde suas relações entre massa e carga (m/z) são medidas. Dentro do instrumento, os peptídeos também podem ser ainda mais fragmentados, produzindo padrões de fragmentação característicos que fornecem informações estruturais sobre os peptídeos. Ao analisar estes padrões, as linhas são representadas graficamente no gráfico espectral m/z permitindo a determinação da sequência de aminoácidos dos péptidos. Esta informação é então comparada com bancos de dados para identificar as proteínas originais. No geral, a espectrometria de massa funciona separando íons com base em seus valores m/z e registrando sua abundância, criando um espectro que revela tanto a composição quanto a estrutura dos peptídeos, conforme mostrado na imagem abaixo.

A espectrometria de massa em tandem permite maior fragmentação desses peptídeos, produzindo íons de fragmentos menores que podem revelar a sequência de aminoácidos. O sequenciamento baseado em espectrometria de massa concentra-se principalmente em processos de ionização, vias de fragmentação e detecção de íons fragmentos. A combinação dos quais proporciona uma impressão digital da sequência peptídica. Esta abordagem é altamente eficaz na identificação de modificações pós-traducionais. O sequenciamento de MS também pode lidar com uma gama muito mais ampla de tamanhos e complexidades de proteínas em comparação com a degradação de Edman.
Técnicas e tecnologias modernas
Como o DNA codifica as instruções para a síntese de proteínas, o sequenciamento do DNA muitas vezes pode fornecer as mesmas informações que o sequenciamento de proteínas. O sequenciamento de alto rendimento, frequentemente chamado de sequenciamento de próxima geração (NGS), permite o sequenciamento rápido e paralelo de milhões de fragmentos de DNA. O processo começa com a fragmentação do DNA, seguida pela ligação do adaptador e amplificação através de técnicas como amplificação em ponte ou PCR em emulsão. Os nucleotídeos são incorporados às cadeias de DNA em crescimento, com processos químicos como pirosequenciamento ou química de terminador reversível detectando a sequência em tempo actual. Os avanços na automação aumentaram significativamente o rendimento, reduzindo custos e tempo e gerando conjuntos de dados massivos. Às vezes, porém, os pesquisadores podem preferir sequenciar proteínas em vez de DNA, a fim de estudar modificações pós-traducionais, que não podem ser determinadas apenas a partir da sequência de DNA. Além disso, o genoma do organismo pode ser desconhecido ou incompleto. Nestes casos, os cientistas voltam aos métodos tradicionais de sequenciamento de proteínas.

Outra técnica moderna de sequenciamento de proteínas é o sequenciamento de proteínas nanopore. Este método analisa moléculas individuais, guiando-as através de um poro em nanoescala (nanoporo) embutido na membrana. À medida que a molécula passa, uma corrente elétrica flui através da membrana, criando interrupções mensuráveis. Para proteínas, o sequenciamento de nanoporos detecta alterações atuais com base na sequência de aminoácidos, permitindo a análise de proteínas em tempo actual. O nanoporo, muitas vezes feito de proteínas biológicas ou materiais sintéticos, forma um canal altamente sensível. Cada aminoácido ou modificação causa uma mudança única na corrente, que algoritmos de computador avançados decodificam. Este método pode sequenciar proteínas em estados nativos ou desnaturados sem preparação extensa de amostras, very best para sequenciamento de alto rendimento.
Aplicações de sequenciamento de proteínas
O sequenciamento de proteínas traz aplicações vitais no mundo actual na medicina, na biotecnologia e até na agricultura. Na área médica, o sequenciamento de proteínas auxilia na identificação de doenças genéticas e mutações, além de desempenhar um papel basic no desenvolvimento de vacinas. Nas indústrias farmacêuticas, o sequenciamento de proteínas auxilia no projeto drogas direcionadas e terapias para doenças. Além disso, na agricultura, melhora a resistência das culturas a pragas e doenças. O sequenciamento de proteínas também auxilia na produção de biocombustíveis e bioplásticos no campo da biologia sintética. Além disso, apoia a ciência forense através de métodos de identificação baseados em proteínas. O potencial para sequenciamento de proteínas na ciência é aparentemente ilimitado.
Limitações do sequenciamento de proteínas
As técnicas de sequenciamento de proteínas enfrentam limitações de precisão e eficiência devido à complexidade e diversidade das estruturas proteicas. Existe uma dependência de amostras de alta qualidade que podem dificultar o sucesso, uma vez que amostras degradadas ou impuras muitas vezes produzem sequências incompletas. Os métodos atuais lutam para identificar modificações pós-traducionais, que desempenham papéis cruciais na função proteica. Muitas técnicas também requerem grandes quantidades de proteína, tornando a análise desafiadora para proteínas de baixa abundância. Além disso, o sequenciamento de proteínas pode ser demorado e caro, o que limita sua acessibilidade.
A espectrometria de massa muitas vezes tem dificuldade em identificar proteínas com pesos moleculares semelhantes. Portanto, a MS nem sempre detecta variações pequenas ou sutis na composição de aminoácidos. A degradação de Edman, por outro lado, é incapaz de sequenciar proteínas além de um determinado comprimento. Os métodos de fragmentação podem introduzir erros de sequenciamento, especialmente para proteínas complexas ou altamente semelhantes. O sequenciamento de nanoporos tem problemas de sensibilidade a modificações, nas quais modificações complexas podem passar despercebidas ou ser mal interpretadas. No geral, os métodos atuais de sequenciamento carecem de sensibilidade para uma análise abrangente de proteomas complexos.




")
